Snakemake Workflow Steps
This document describes the purpose of the individual steps in the Snakemake workflow. Note that certain model evaluation and figure-generation steps have been omitted from this discussion.
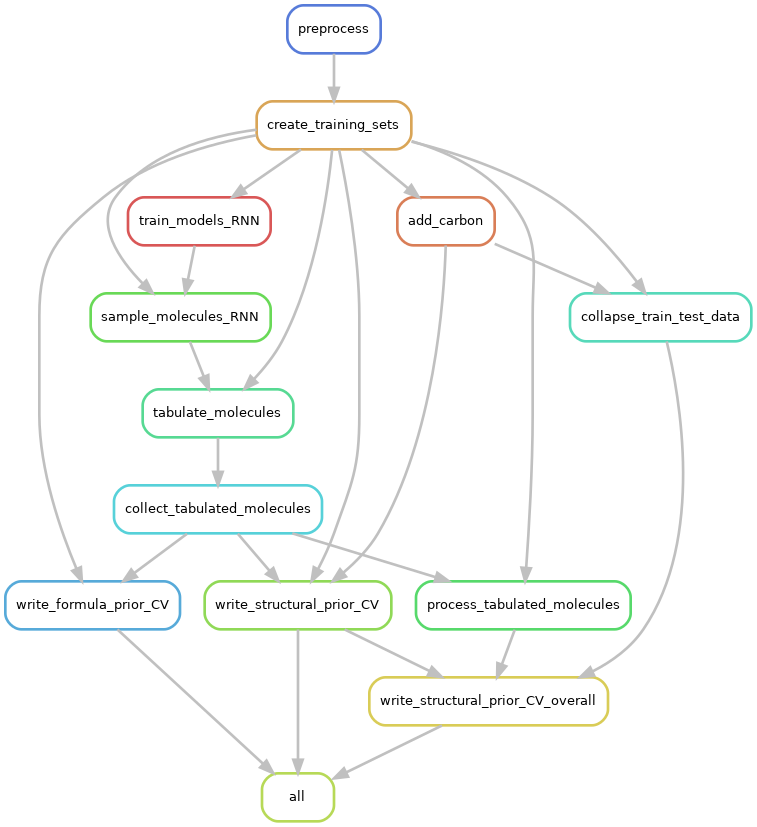
preprocess
Read input smiles and save a file of “canonical” smiles. This entails conversion
to molecules using rdkit, removing light fragments, neutralizing charges,
filtering for valid elements, removing very rare tokens in the smiles vocabulary (and eliminating the corresponding molecules), and saving the resulting smiles.
Inchikeys are added to each individual smile, and any extra metadata for an input smile is saved in the output file.
create_training_sets
Split input smiles into separate files for train/test folds, while also creating
.vocabulary files for the training smiles. The number of folds can be specified in the config.yaml file.
The smiles can optionally be (and typically are) augmented
by an enum factor (typically 10x to 100x) to increase the size of the training set, and subsequent
steps of the pipeline are run for each enum factor in separate folders.
When creating training sets, the prefix train_ is used for files that might possibly be augmented, and train0_ for the non-augmented files. The former are useful
for training purposes, while the latter are useful for evaluation purposes. Similarly, the prefix test_ or test0_ is used for the test files.
This step also optionally takes in a min_tc parameter to only consider smiles that are mutually similar (to a randomly selected seed smile).
add_carbon
This step generates data for the AddCarbon model by inserting a
random carbon atom at a random point in training set SMILES. This is useful for model evaluation purposes later on (in the step write_structural_prior_CV).
collapse_train_test_data
This step generates single files for training and testing smiles respectively, out of all the folds we generated in the create_training_sets step. This is useful
for the write_structural_prior_CV_overall step, explained later.
train_models_RNN
This step trains one or more (the number of train_seeds entries in config.yaml) RNN models on an input fold. This is the most time and resource intensive step
in the entire pipeline. The specifics of the model (number of layers/epochs/batch size etc). can be configured in the model_params section of the config.yaml file.
Each training fold is internally split into train/validation split of 0.9. The trained model and loss files are written out.
sample_molecules_RNN
This step samples sample_mols (typically of the order of 10M) smiles from a trained model and fold, and saves to and output file. It is possible to sample multiple times
from the same trained model using multiple sample_seed values.
tabulate_molecules
For sampled smiles from a model and a particular fold, this step adds the calculated molecular mass, formula and sampling frequency. Smiles that are found in training file for that particular fold are filtered out, based on a comparison of InchiKeys.
collect_tabulated_molecules
This is an aggregation step for all sampled smiles from all samples obtained across all |train_seeds| models in a fold,
and adds aggregation metrics like sampling frequency.
write_structural_prior_CV
This step evaluates test smiles against the trained models for each fold, with the PubChem and AddCarbon model as baselines.
For each fold, for each smile in the test dataset, we generate statistics for the occurrence of the test smile in each of the 3 “models” (trained_smiles/sampled_smiles/PubChem).
For each of the 3 “models”, we keep track of smiles that fall within some tolerance of the true test-smile molecular weight. When sorted by decreasing
sampling frequency, the rank at which we find the “correct” smile (in terms of a match in the smile string), gives us the “rank” of each test smile.
Lower ranks indicate a better model. This output is written to ranks_file.
write_formula_prior_CV
This step is analogous to write_structural_prior_CV, but here “correctness” is based on a matching formula, not a matching smile.
process_tabulated_molecules
This step aggregates sampled smiles across all folds, calculating metrics like average sampling frequency (if metric=freq-avg).
write_structural_prior_CV_overall
This step is analogous to write_structural_prior_CV, but evaluates test smiles against the trained models across all folds.